Wiki
Clone wikiATLAS / Home
Welcome to ATLAS, your guide to the world of low-depth and ancient DNA!
ATLAS stands for Analysis Tools for Low-coverage and Ancient Samples. These tools cover all programs necessary to obtain variant calls, estimates of heterozygosity and more from a BAM file. There are sequence data processing tools, diagnostic tools, and variant discovery tools, similar to GATK by the Broad Institute.
For instructions on how to install and run ATLAS click here.
For news and version release notes click here.
Each page of this wiki describes a different "task" implemented in ATLAS. Engine parameters that are common to all tasks can be found here.
Standard Pipelines
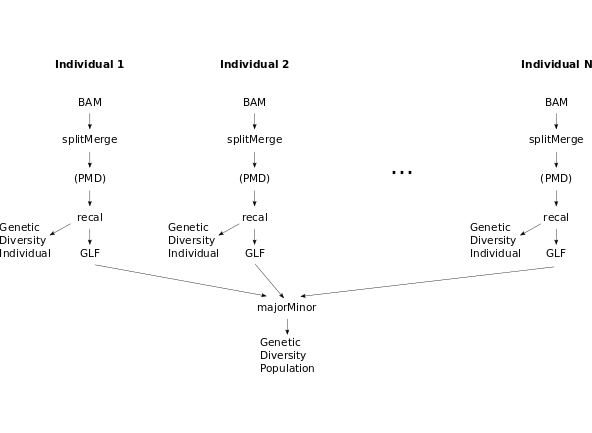
We highly recommend using a local realignment tool on your BAM file and validating it with Picard-tools validateSamFile before taking off with ATLAS.
Standard pipeline for the analysis of individual samples:
- splitMerge: Split the single-end read groups according to length and merge the mates in paired-end read groups. This creates a new BAM file, which should be used in the subsequent steps.
- PMD: If your sample is ancient, estimate the post-mortem damage patterns. Otherwise, skip this step.
- recal (or BQSR): Recalibrate the base quality scores.
Steps 2. and 3. produce files containing PMD and base quality score parameters, which ATLAS uses to calculate accurate genotype likelihoods. They should be provided in subsequent quantification of genetic diversity.
- Infer the individual genetic diversity parameter of interest
Standard pipeline for the analysis of population samples:
- Follow steps 1-3 of the standard pipeline for the analysis of individual samples
- GLF: For every sample, write the genotype likelihoods to GLF file (provide the PMD and recalibration parameters)
- major/minor: Find the two most likely alleles in the population for every locus and write the genotype likelihoods for these alleles for every individual into a VCF file
- Infer the population genetic diversity parameter of interest
All ATLAS Tasks
Base Quality Score Recalibration
Post-Mortem Damage
Genetic Diversity in Individuals
Genetic Diversity in Populations
- create GLF file
- printGLF
- major/minor
- genetic distance between individuals
- inbreeding coefficient F
- estimate allele counts
- estimate allele frequencies
- calculate F2
BAM Diagnostics
- BAMDiagnostics : Overall depth, Mapping quality, read length and fragment length distributions
- readOverlap
- qualityDist
- depthPerSiteDist
- assess soft clipping
- writeDepthPerWindow
- writeDepthPerSite
- pileup
- createDepthMask
BAM Manipulation
- splitMerge
- mergeReads (deprecated, use splitMerge)
- splitRGbyLength (deprecated, use splitMerge)
- BAMUpdateQualities
- binQualityScores
- mergeReadGroups (deprecated, use poolReadGroups during recalibration or PMD estimation)
- downsample
- downsampleReads
VCF Diagnostics
- VCFCompare
- VCFToInvariantBed
- VCFAssessAllelicBalance
- polymorphicWindows
VCF Manipulation
- VCFFixInt
VCF Format Transformation
- VCFToBeagle
- VCFToLFMM
- VCFToPosFile
Other
Tutorials
SAM flag requirements
In all tasks, reads that are not tagged as a "proper pair" by the SAM flag are considered to be single-end.
Disclaimer
ATLAS is under active construction and although we have a test suite we do not guarantee that our code is bug-free.
Questions?
See our Frequently Asked Questions or write an email to daniel.wegmann@unifr.ch
Updated